





歐盟新規申報難度超FDA,強制公告機構門檻提高!
2017年5月5日,歐洲聯盟(EU)發布了新版醫療器械法規MDR(EU2017/745),以替代舊版醫療器械指令MDD(93/42/EEC)和有源植入醫療器械指令AIMDD(90/385/EEC),同年5月26日生效,過渡期三年。過渡期內,醫療器械廠商可以自主選擇以舊指令MDD或新法規MDR申請CE證書;過渡期結束將強制執行新版MDR法規。

歐盟醫療器械法規歷史進程節點(動脈網制圖)
然而,原計劃于2020年5月26日強制執行的新版MDR法規,由于受到疫情等相關因素影響,被宣告推遲一年,直到今年5月26日才正式強制執行。
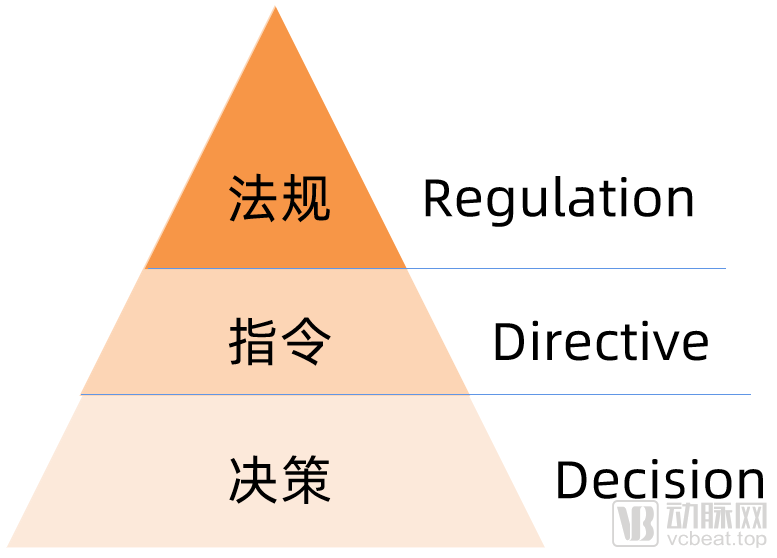
歐盟法規的金字塔體系(約束力由高到低)
從Directive(指令)到Regulation(法規),歐盟提高了對醫療器械的約束力,發布立即在歐盟成員國生效并成為有約束力的法律,此次的Regulation無需像Directive那樣需要經過成員國轉化成當地法律法規再去落地實施。
值得一提的是,在歐盟醫療器械分類當中,將醫療器械劃分為醫療器械(MD)和體外診斷器械(IVD)兩大類,目前受到新規MDR管轄的僅限于MedicalDevices,體外診斷器械的相應新規IVDR(EU2017/746)的執行時間為2022年5月26日,也就意味著IVD器械廠商還有一年的時間可以緩沖和適應新規變化,并做好應對準備工作。
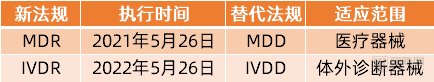
歐盟的兩類醫療器械法規及執行時間
對于其他醫療器械(MD)廠商來說,從2021年5月26日開始,如果要申請歐盟認證則必須遵從新規MDR執行。相對于舊法令MDD,歐盟新法規MDR發生了哪些變化?面對這些變化,國產醫藥器械廠商在申請CE認證時需要注意的點有哪些?對于之前已經申請CE認證的廠商,未來如果繼續持有CE證書需要做好哪些應對工作?
歐盟新規申報難度超FDA,強制公告機構(NB)門檻提高
在過去,歐盟CE認證的難度較中國NMPA及美國FDA更低,背后的原因則是歐盟醫療器械舊法規的約束力松弛,對醫療器械研發商報批要求普遍較低。同時也導致了部分僅獲得CE認證的醫療器械在歐盟地區落地后也常出現醫療事故,所以這些產品未來進入中國及美國市場時,仍然需要面臨更長期、更嚴格的報批流程。
在新規執行之前,一些低風險醫療器械產品研發廠商在申請CE認證時,可以通過自我聲明的方式進行申請,但這種申請的方式監管并不嚴格、缺乏約束力。所以在2020年業內還出現了一則曝光事件,中國深圳某醫療器械公司產品通過自我聲明方式出口歐盟,最終發現90%產品不合格。
如果產品沒辦法精準管控,那么CE認證的“含水量”便較高。據業內人士透露,針對歐盟這種情形,英國、德國甚至還出現過“小名單”——如果你的產品只在歐盟拿了認證,那么該產品在英德申報時還是需要經歷嚴格的臨床試驗,以確保產品的安全性、有效性。
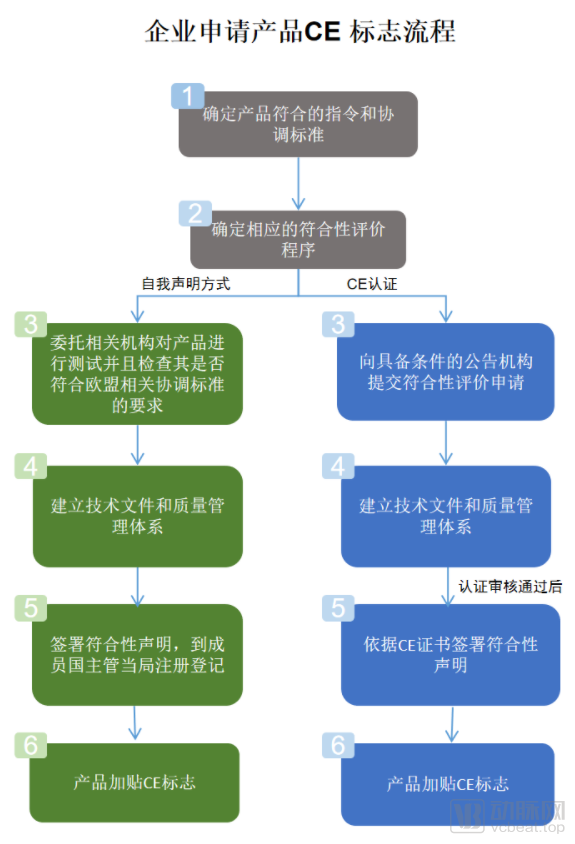
歐盟醫療器械新舊法規申報流程變化(截圖自關務小二&中國國際貿易促進委員會官網)
但在歐盟MDR新規執行之后,即便是一些電商平臺上進行銷售的醫用產品也需要通過嚴格的授權公告機構(Notified Body,NB/歐代)進行申報。與此同時,歐盟對NB機構的要求也大幅提升,公告機構是獨立于進行符合性評估活動的產品制造商的第三方機構,要求長期配備具有相關資格證書/資質的產品審查人員、質量管理體系審核人員等,且不能采取外包機制聘用。
對公告機構(NB)的高要求,也讓目前獲批的能夠進行新規公告的機構數量大幅降低。根據歐盟官網信息查詢可以看到,過去通過MDD授權的公告機構總計有51家。
然而,自今年5月26日起,這些通過MDD指定的公告機構已不能再根據其指令頒發新證書,而只能對之前頒發的有效證書進行監督過渡。據統計,目前通過歐盟新規MDR授權的公共機構僅有20家。

 分享
分享
請輸入評論內容...
請輸入評論/評論長度6~500個字

圖片新聞










