





歐盟新規申報難度超FDA,強制公告機構門檻提高!
2、 專門用于與中樞神經系統直接接觸的醫療器械。
3、 用于管理醫藥產品的植入式器械及長期手術侵入性器械。
4、 有源植入式器械或其附件。
5、 乳房植入物或手術網。
6、 全部或部分關節置換物(輔助部件如螺釘、楔子、板等除外)。
7、 椎間盤置換植入物或與脊柱接觸的可植入裝置(如上,輔助部件除外)。
8、 所有旨在控制、監測或直接影響有源植入式設備性能的有源設備。
9、 提供用于診斷或治療目的決策軟件,其決策可能導致人體死亡或健康狀況產生不可逆轉的惡化。
10、 具有高等、中等內照射潛力的納米材料組成的設備。
11、 通過身體孔口引入人體或應用于皮膚并被人體吸收或局部分散的物質及物質組合組成的裝置(包括物質及其代謝物被人體吸收達到預期目的/物質及其代謝物在胃或消化道被人體吸收達到預期目的)。
12、 具有集成或合并診斷功能的有源治療設備,如閉環系統或自動體外除顫器。

MDR相對于MDD分類細化部分
從這次歐盟MDR新規新增的部分我們可以看出,MDR新規在分類上對舊MDD法令進行了進一步細化,對于一些原本模糊的分類進行了邊界清晰界定,以及根據同類產品可能帶來的不同后果嚴重程度進行了等級劃分。
例如,在對決策軟件的分類上,其決策如果可能導致人體死亡或健康狀況不可逆轉的惡化,該軟件則被劃分為最嚴格的Ⅲ類;如果其決策可能導致一個人的健康狀況或手術干預嚴重惡化,該軟件則被歸類為Ⅱb類;而其余旨在提供用于診斷、治療、監測生理過程目的的決策軟件都被歸類為IIa類。
同樣,MDR也新增了對包含或由納米材料組成的設備的分級歸類:如果其設備呈現高或中等內照射(internal exposure)潛力,責備認定為Ⅲ類;如果是較低的內照射可能性,則歸為Ⅱb類;如果其呈現出的內照射潛力可以可忽略不計,則歸為Ⅱa類。
依據歐盟分類的原則,如果該器械或者軟件對患者存在更大的潛在影響,都被歸類為更嚴格的管控等級。如MDR法規中第20條分類規則,將除外科手術侵入性器械外,所有與身體孔道相關的、旨在通過吸入給藥的侵入性器械均歸為Ⅱa類,但是如果該器械作用方式對給藥的藥物有效性和安全性具有重要影響或它們旨在治療危及生命的疾病,在這種情況下,則被歸類為Ⅱb類。
同時,因為MDR新規重新定義了臨床數據的種類和可用性,針對數字健康應用(digital health APP)的醫療器械危險等級分類也會帶來重要的區別。目前已經通過德國DVA審核的數字健康應用(DiGA), 需要在基于MDR新規下,確認其產品風險是否升級。如果該APP已經不再滿足DVA法案所規定的Ⅰ或 Ⅱa類,升級到Ⅱb類甚至更高危險等級分類,其將不再是DiGA。
醫療器械產品溯源:增設醫療器械的數據庫、唯一器械標識(UID)
除了對醫療器械基礎的定義和分類進行了進一步完善,MDR與MDD的還有一大優化在于,歐盟MDR新規要求建立歐洲醫療器械數據庫(Eudamed)和對應的唯一器械標識(UID),方便公眾充分了解醫療器械相關信息、并實現設備的逐一溯源調查。
醫療器械數據庫(Eudamed)需要包含7大電子系統:器械注冊電子系統、UDI數據庫、經濟運營商電子登記系統、認證機構和證書電子系統、臨床研究電子系統、警戒和上市后監管電子系統、市場監管電子系統。
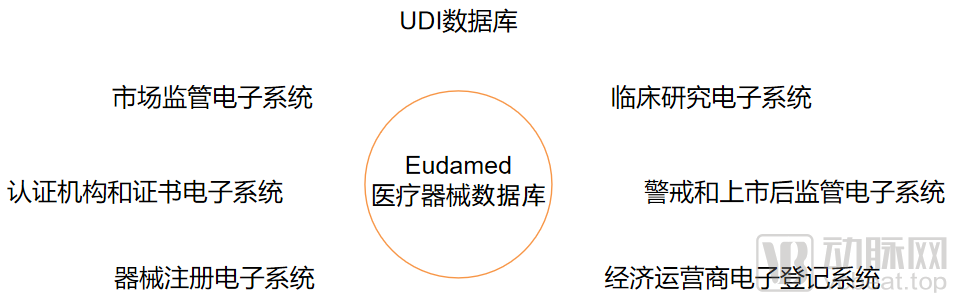
醫療器械數據庫的組成系統(動脈網制圖)
這七大電子系統的數據由成員國、認證機構、經濟運營商和申辦方錄入,Eudamed進行整理并加工所有信息,最后向認證機構、經濟運營商、申辦方和公眾開放信息訪問權限。其中,對于Ⅲ類器械和植入式器械(非定制或研究器械),制造商應起草一份安全和臨床性能總結,該總結需要通過Eudamed向公眾開放。
這里值得一提的是需要被錄入到醫療器械數據庫中的UDI數據,UDI數據相當于醫療器械的“身份證”,由UDI-DI和UDI-PI兩部分組成。

醫療器械“身份證”UDI組成(動脈網制圖)
UDI-DI可以理解為“產品識別碼”,由企業識別碼和產品規格碼兩部分組成,是UDI中固定且強制性要求的部分,需要上傳到UDI編碼數據庫向公眾公開;而UDI-PI則是器械的“生產識別碼”,由醫療器械序列號、生產批號、生產日期、失效日期等生產相關信息組成,非必要性也無需上傳到UDI編碼數據庫。
從全球范圍來看,2008年,國際醫療器械監管機構論壇(IMDRF)建立UDI特別工作組,并于2011年9月通過了《醫療器械唯一標識系統指南》;2012年,IMDRF繼續補充完善通過了《醫療器械唯一標識系統擬定規則》,指導全球各監管部門依照該擬定規則來構建自己的UDI系統。
目前,在IMDRF的10個國家和地區中,美國和歐盟已經發布了UDI相關法規,不過美國FDA的UDI是由貼標人負責,歐盟的則由制造商負責,且對貼標語言有一定要求,必須是歐盟成員國的官網語言。另外,巴西、中國和韓國也陸續發布了法規的征求意見稿,日本、加拿大和澳大利亞發布了UDI相關通知和指導性文件。醫療器械的全面UID時代即將到來!
加強上市后監管體系:提高醫療器械性能、臨床安全性要求
在歐盟新規發布之前,依據歐盟舊法MDD報批CE認證難度較低,所以國內不乏一些醫療器械廠商其產品已經在海外通過CE認證,但是在國內卻遲遲沒有拿下上市許可的情況。
不過此次歐盟MDR新規強制執行之后,將大大提升CE認證的難度,同時提高了CE認證的含金量。在新規中,歐盟強調了提高醫療器械安全性、性能及相關文件要求的重要性,并且加強了器械上市后的監管體系。
在MDR附錄Ⅰ第一章一般要求中提出,制造商應建立、實施、記錄和維護風險管理體系,其中風險管理應理解為在器械整個生命周期中為連續迭代過程,需定期進行系統更新。并且制造商還需要起草一份包含UDI-DI等8個方面內容在內的安全及臨床性能總結給到CE公告機構,并進行驗證。后續總結報告還會被上傳到Eudamed數據庫中接受監管。
此外,MDR中添加了對申報技術文件內容的要求,且明確指出上市后監管計劃和安全性更新報告(PSUR)都是技術文件的一部分,并要求依據上市后監管體系收集的資料對技術文件中相應信息進行更新。
醫療器械制造商應計劃、建立、記錄、實施、維護和更新上市后的監管體系,上市后的監管體系應適于積極和系統地收集、記錄并分析器械在其整個生命周期內的質量、性能和安全相關數據,以得出必要的結論,并確定、實施和監測任何預防及糾正措施。
在PSUR上,Ⅱa、Ⅱb和Ⅲ類器械制造商應針對各器械或類別或器械組編制定期安全性更新報告(PSUR),總結數據分析結果和結論,并對采取的任何預防和糾正措施提供理由和說明。其中,Ⅱb和Ⅲ類器械的制造商應至少每年更新PSUR;Ⅱa類器械制造商應在必要時至少每兩年更新PSUR。
最后,歐盟新規MDR還豐富了對醫療器械審核的臨床評價部分,設立了專門的專家小組以評估審查制造商報批醫療器械的預期臨床用途和臨床研究方案,制造商應適當考慮專家小組發表的意見,但不得對專家小組就任何未來合格評定程序所表達的觀點行使任何權利。
歐盟MDR新規下的影響:30%中小器械企業及OEM面臨倒閉風險
通過分析整個歐盟MDR新規,不難發現歐盟新規的完善一定程度上參考了國際上成熟的醫療器械法規,特別是美國FDA對醫療器械的審批要求,這也讓全世界達成一致的醫療器械規范更進一步。
“嚴格的法規對整個醫療行業健康發展的影響總體上是積極的。”體外診斷上游醫療器械研發企業匯先醫療CEO顏菁認可道,“盡管歐盟MDR新規的實施將給中國出口企業帶來成本增加、認證周期拉長和合規風險增加等問題,但法規對促進醫療器械廠商以更嚴格的標準要求自己,保障產品和標準的符合性,這對患者而言都是有利無害的,對整個醫療器械行業既是挑戰,也是機遇。”
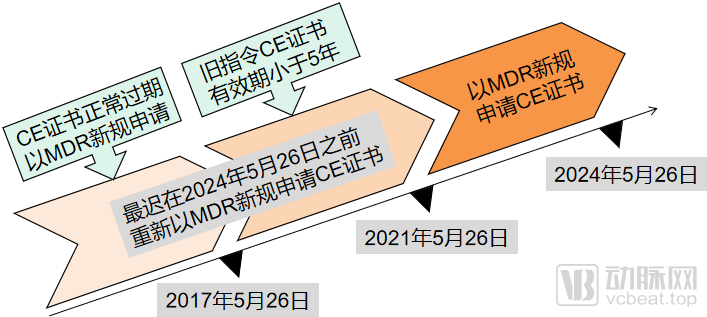
不同時期拿到CE證書企業的注意事項(動脈網制圖)
對于在過渡期內依據舊指令MDD、AIMDD或IVDD公告機構簽發的CE證書將在有效期內繼續有效,但是從其交付日期起有效期不得超過5年,并且最遲于2024年5月26日失效。
而對于在過渡期之前獲得的CE證書已經上市的醫療器械產品,需要在舊CE證書過期/失效之前(同樣最遲于2021年5月26日),盡快重新申請基于MDR新規的CE證書:確認其產品風險分級是否升級,以及確認原CE證書的發證機構是否還具備MDR授權公告的資格,盡快修改原CE技術文件,再次向具有MDR發證資質的NB機構提出新的認證申請。
這也就意味著2024年之后,整個歐盟市場將不再存在基于舊法規的CE證書,醫療器械研發商盡快適應新規是當務之急,“提早做好迎接新規準備的醫療器械研發商在未來收入將會增加,而那些歐盟本土20%-30%的中小醫療器械廠商將會面臨出局的局面。”3D打印醫療器械公司智塑健康CEO張靖表示。
與此同時,由于歐盟申請門檻的提高,以及對醫療器械研發過程的監管,國內傳統通過OEM代工生產再到歐盟進行產品CE認證的路徑也將徹底行不通,以代工生產醫療器械為核心業務的企業也將被迫轉型。但這對于整個醫療器械歐盟市場的發展無疑是重大利好的,歐盟是僅次于美國的全球第二大市場醫療器械市場,占比全球醫療器械市場總和的26%,所以維護醫療器械企業的市場準入至關重要。
作者:王嬋

 分享
分享
請輸入評論內容...
請輸入評論/評論長度6~500個字

圖片新聞










